nfcore introduction
![]()
Nf-core is a very active community around nextflow. Volunteers develop nextflow pipelines around a variety of bioinformatic data.
Here are some flagship pipelines that have been developed by the nf-core community (we will have a look at the entire list on the nf-core homepage in a bit):
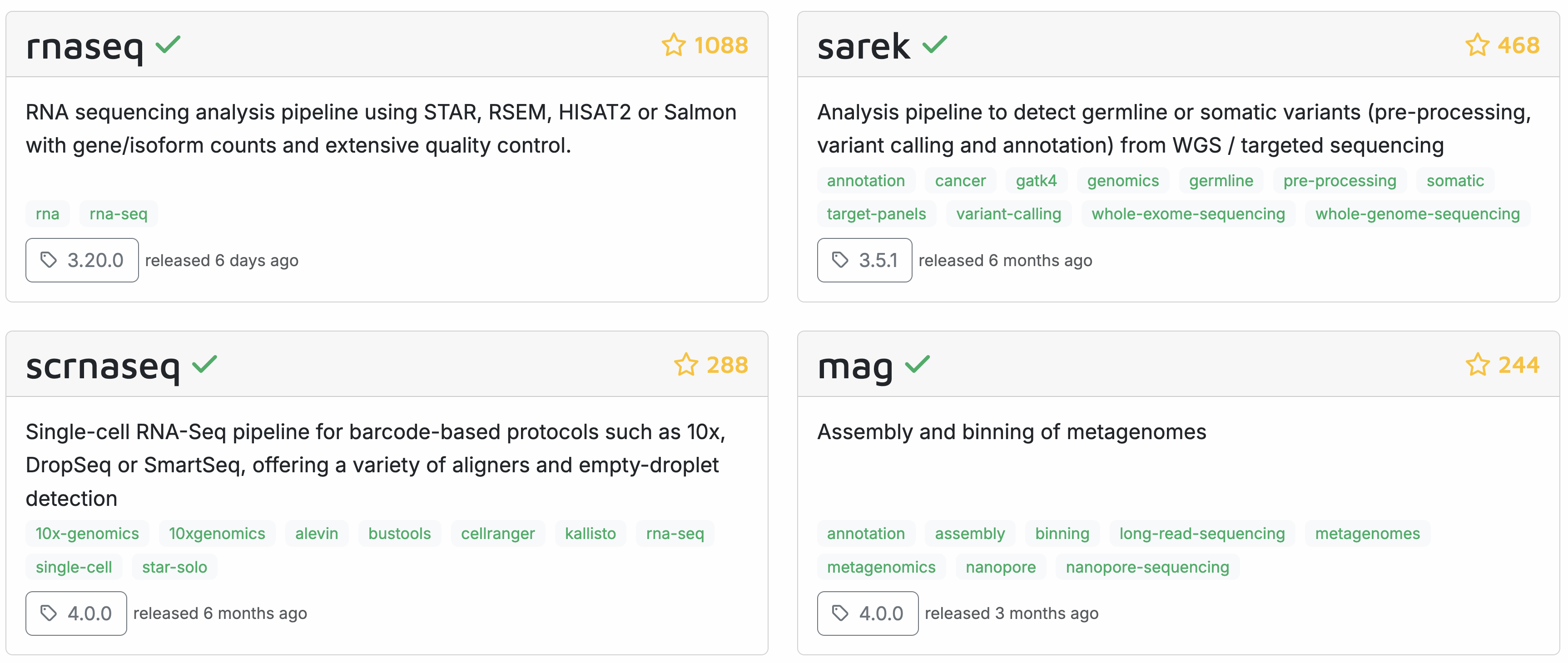
All nf-core pipelines are open source and the source code is available on github. The pipelines are developed by volunteers, who can have a very varied background.
While nf-core is fantastic, please be aware that their pipelines are developed and maintained (or not maintained) by the community. You should not use the pipelines as a black box, but as a tool you need to understand. The responsibility for the end results is still yours, so you need to see if your data is suited for the analysis (good enough quality?), and if the analysis is suitable for your data!
However, nf-core does not only develop pipelines.
The community also develops:
- processes that they make available as
modules(and optimizes them too). - training material for all user classes.
- best practices for documentation.
- templates for development.
There is a weekly online helpdesk, and even a podcast.
What makes nf-core pipelines interesting for you?
Nf-core provides already developed pipelines for many different data sets. Likely, a pipelines exists that you can use on your data. The documentation of the pipelines follows nf-core guidelines and is extensive and informative - it is easy to understand what the pipeline does and how it works. All output is explained in detail, with links to more extensive documentation.
Once you have understood how to run a nf-core pipeline their consistency and standardization means you will know most about running a different one.
Using the nf-core launcher, will check your input, and automatically generate commands and configuration files.
And this is on top of all nextflow functionality such as portability, reproducibility and the resume-at-fault option!
Finding and evaluating a pipeline
From the nf-core homepage, we can search for pipelines. We are going to demo the rnaseq pipeline. You can see that there is a lot going on in this pipeline! We will chat more about these things in class rather than including screenshots of everything.
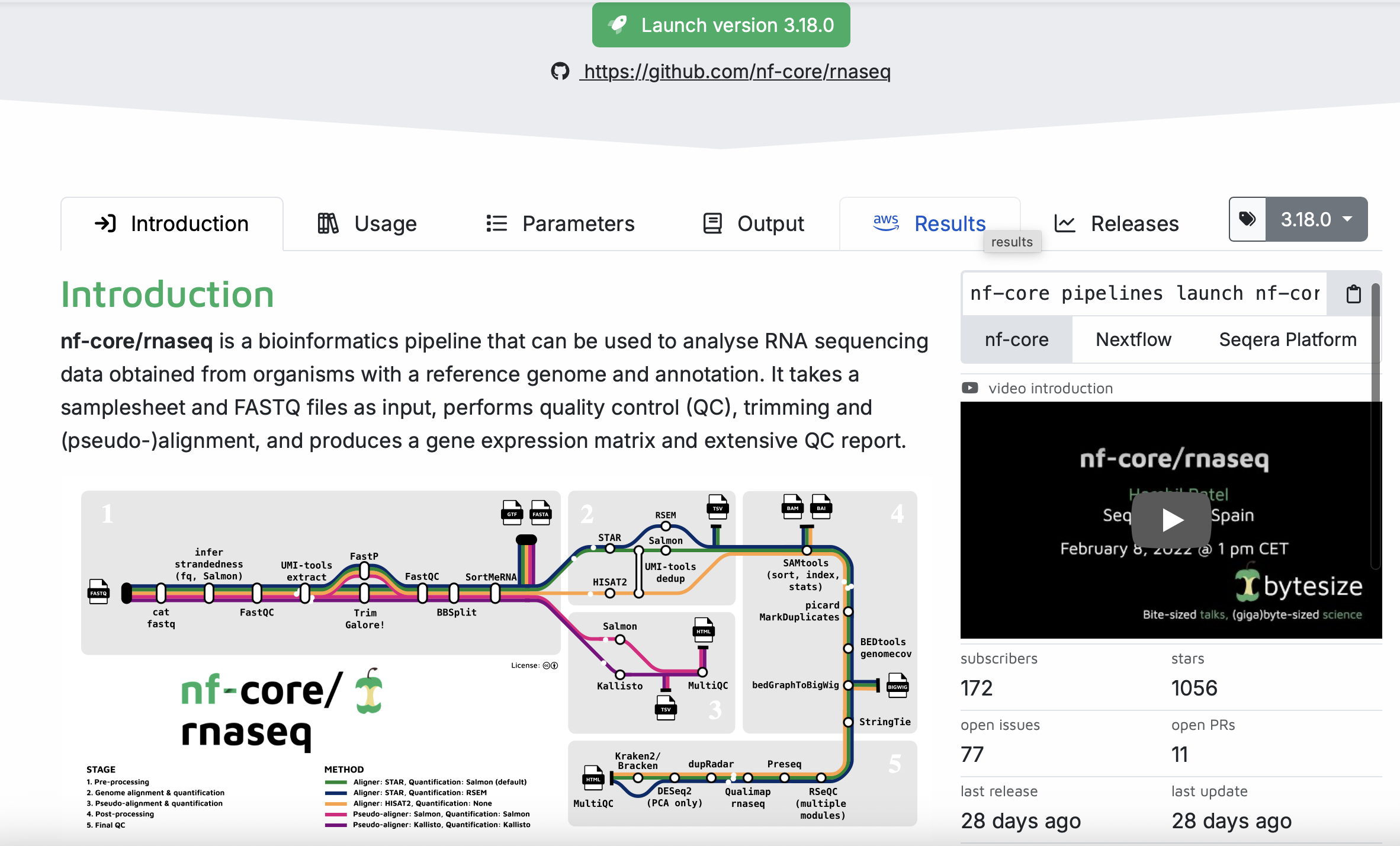
Under the Usage, you will find a lot of information that describes the pipeline, including input information, the samplesheet.csv (which we will have to set up with our data later).
Under the Parameters tab you will find information on all of the things that we will set up in the next step.
Under the Output tab, you will find information on the expected output generated from the pipeline. This is useful to help you interpret what the pipeline produces.
Look through the available pipelines and see if there is one that is interesting for you and your project.
In the next part we will set up a nf-core pipeline with the test profile, so then you can test the pipeline that is most interesting for you.