Nextflow
When developing your code in bioinformatics, you will likely use different tool for different parts of your analyses. Traditionally, you would have about one script per tool, all of which you deploy by hand, one after the other. Together, this is called a workflowor pipeline.
Manual deployment of pipelines can be tedious, especially if you have analyses with many steps, or many samples of different sizes that might need a varying amount of computational power. Luckily for you, other bioinformaticians and software developers have developed something to make your life much easier:
Workflow managers
Workflow managers provide a framework for the creation, execution, and monitoring of pipeline. <…> They simplify pipeline development, optimize resource usage, handle software installation and versions, and run on different compute platforms, enabling workflow portability and sharing. Wratten et al. (2021) Nature Methods
With workflow managers you can develop an automated pipeline from your scripts that can then be run on a variety of systems. Once it is developed, execute a single command to start the pipeline. The manager then coordinates the deployment of the scripts in the appropriate sequence, monitors the jobs, handles the file transfers between scripts, gathers the output, and handles re-execution of failed jobs for you. Workflow managed pipelines can run containers, which eliminates software installation and version conflicts.
That means that by design the pipelines are:
- portable
- more time efficient (no more downtime between pipeline steps)
- more resource efficient (mostly, but this might vary depending how skilled a developer you yourself are)
- easier to install (especially when combined with containers, or environment managers)
- more reproducible
There are in principle two different flavors of workflow managers: snakemake, and nextflow. In this course we will be introducing you to nexflow.
Nextflow
In nextflow, your scripts are turned into processes, connected by channels that contain the data - input, output etc. The order of the processes, and their interaction with each other, is specfied in the workflow scope.
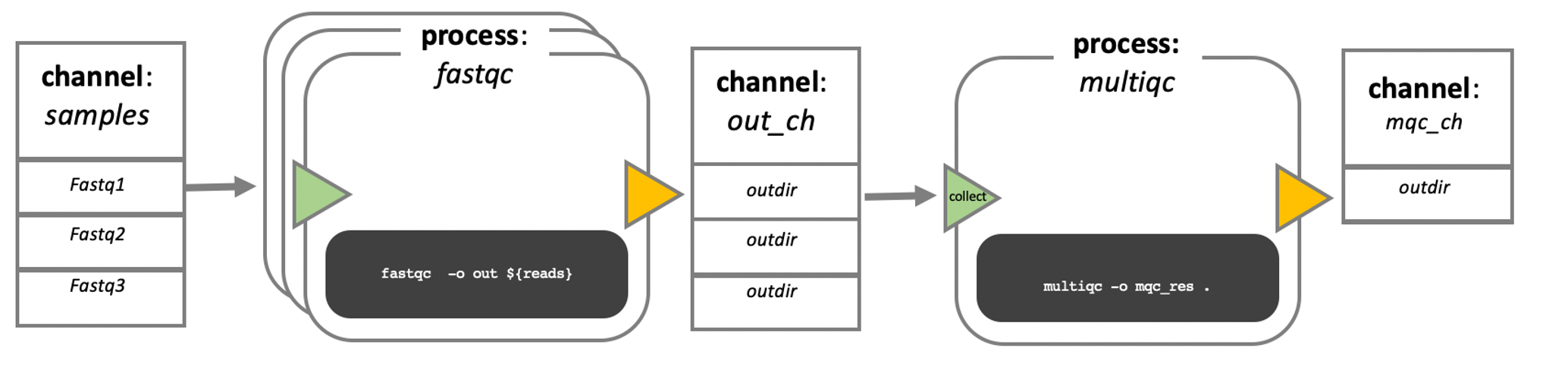
In this course, we will not write our own processes, or pipeline. However, if you are interested, there are a lot of very good training materials available online.
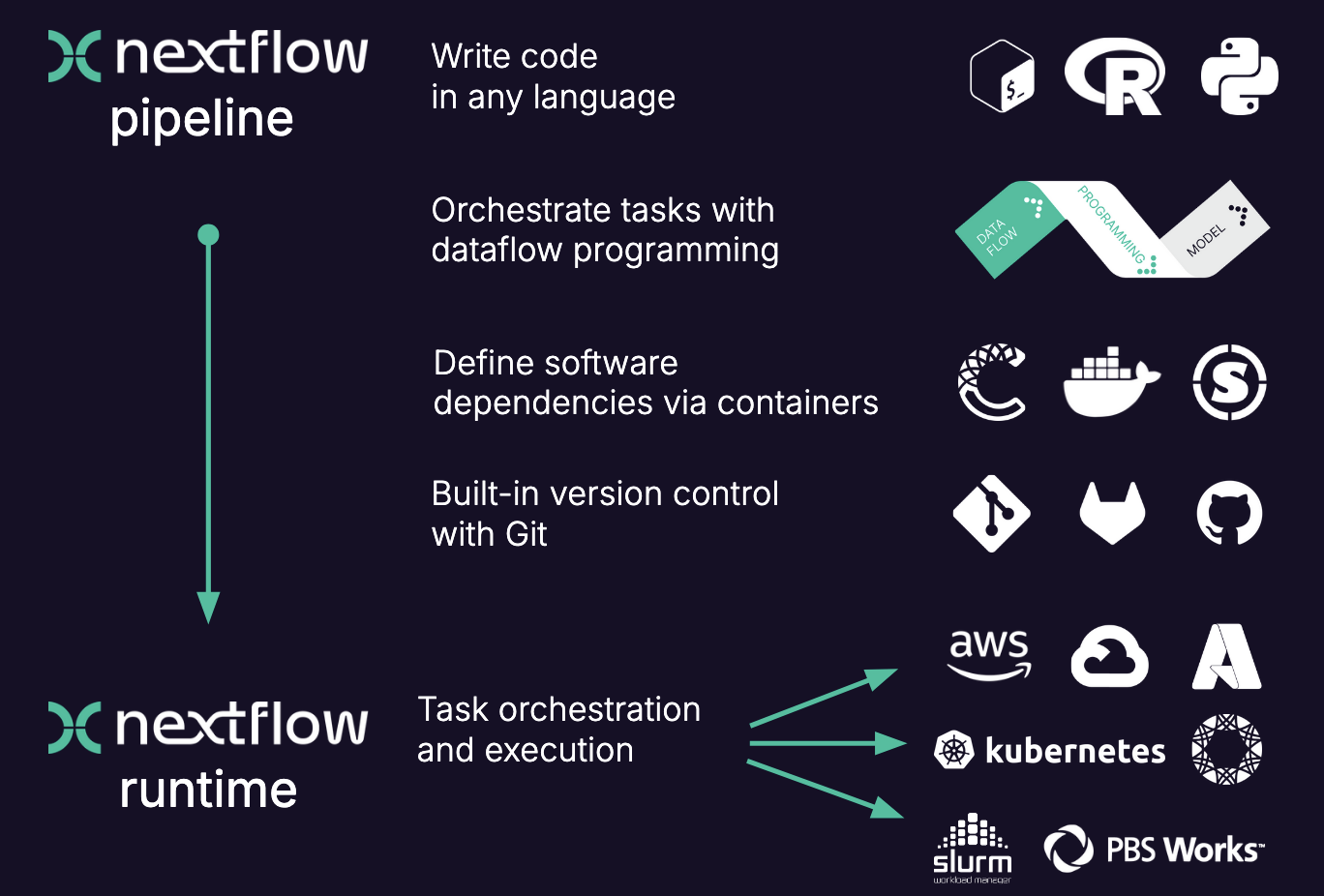
The executable part of the processes, the so called script, can be written in any language, so in theory you could always choose the language that is best suited for the job (in practice you might be limited to the languages you know). However, the modularity of the processes allows for easy re-use of existing scripts and processes.
When moving the pipeline from one system to another, the script stays the same and does not change, the same containers are used. The only thing that changes are the parameters that pertain to the environment and available resources. In other words, with nextflow, the functional logic, the processes, are separated from the executive (how the workflow runs). This makes the nextflow pipelines highly interoperable and portable. They can be run on various platforms, such as HPC clusters, local computers, cloud systems etc..
The pipelines can be integrated with version control tools, such as git or bitbucket, and containers technologies, such as apptainer or docker. This makes the pipeline very reproducible.
The nextflow pipelines are extremely scalable, can be developed on a few samples and easily be run on hundreds or thousands of samples. When possible, processes are run in parallel automatically.
Nexflow performs automatic checks on the processes and their in- and output. It can automatically resume execution at a point of failure without having to re-compute successfully completed parts.
Nextflow is open source.
Here is a more visual summary of some of the points above: